Single-cell Immune Repertoire Trajectory Analysis Starting From scRepertoire
2025-01-13
vignette_from_scRepertoire.Rmd![]()
![]()
![]()
Foreword
Welcome to dandelionR!
dandelionR is an R package for performing single-cell
immune repertoire trajectory analysis, based on the original python
implementation in dandelion.
It provides all the necessary tools to interface with scRepertoire and a custom implementation of absorbing markov chain for pseudotime inference, inspired based on the palantir python package.
This is a work in progress, so please feel free to open an issue if you encounter any problems or have any suggestions for improvement.
Installation
You can install dandelionR from GitHub with:
if (!requireNamespace("devtools", quietly = TRUE)) {
install.packages("devtools")
}
if (!requireNamespace("BiocManager", quietly = TRUE)) {
install.packages("BiocManager")
}
if (!requireNamespace("scater", quietly = TRUE)) { # only for the tutorial
BiocManager::install("scater")
}
devtools::install_github("tuonglab/dandelionR", dependencies = TRUE)In a standard analysis workflow in R, users probably choose to read
in their VDJ data with scRepertoire.
In this vignette, we will demonstrate how to perform TCR trajectory
analysis starting from ‘raw’ data i.e. just a standard single-cell gene
expression data (stored in SingleCellExperiment) and VDJ
data (in AIRR format).
Install scRepertoire if you haven’t already.
if (!requireNamespace("scRepertoire", quietly = TRUE)) { # only for the tutorial
BiocManager::install("scRepertoire")
}
# or
devtools::install_github("ncborcherding/scRepertoire")Usage
Load the demo data
Due to size limitations of the package, we have provided a very trimmed down version of the demo data to ~2000 cells. The full dataset can be found here accordingly: GEX - https://developmental.cellatlas.io/fetal-immune (Lymphoid Cells) and VDJ - https://github.com/zktuong/dandelion-demo-files/tree/master/dandelion_manuscript/data/dandelion-remap
Check out the other vignette for an example dataset that starts from
the original dandelion output associated with the original
manuscript.
Use scRepertoire to load the VDJ data
For the trajectory analysis work here, we are focusing on the main
productive TCR chains. Therefore we will flag
filterMulti = TRUE, which will keep the selection of the 2
corresponding chains with the highest expression for a single barcode.
For more details, refer to scRepertoire’s documentation.
Note: There is a known issue with
combineTCR shuffling the order of the
SingleCellExperiment colData and this may not
have been updated in the latest version of scRepertoire on
Bioconductor (release 3.20). If you encounter this issue, please install
the development version of scRepertoire from GitHub:
devtools::install_github("ncborcherding/scRepertoire")
contig.list <- loadContigs(input = demo_airr, format = "AIRR")
# Format to `scRepertoire`'s requirements and some light filtering
combined.TCR <- combineTCR(contig.list,
removeNA = TRUE,
removeMulti = FALSE,
filterMulti = TRUE
)Merging VDJ data with gene expression data
Next we will combine the gene expression data with the VDJ data to
create a SingleCellExperiment object.
sce <- combineExpression(combined.TCR, demo_sce)Initiate dandelionR workflow
Here, the data is ready to be used for the pseudobulk and trajectory
analysis workflow in dandelionR.
Because this is a alpha-beta TCR data, we will set the
mode_option to “abT”. This will append abT to
the relevant columns holding the VDJ gene information. If you are going
to try other types of VDJ data e.g. BCR, you should set
mode_option to “B” instead. And this argument should be
consistently set with the vdjPseudobulk function later.
Since the TCR data is already filtered for productive chains in
combineTCR, we will set
already.productive = TRUE and can keep
allowed_chain_status as NULL.
We will also subset the data to only include the main T-cell types: CD8+T, CD4+T, ABT(ENTRY), DP(P)_T, DP(Q)_T.
sce <- setupVdjPseudobulk(sce,
mode_option = "abT",
already.productive = TRUE,
subsetby = "anno_lvl_2_final_clean",
groups = c("CD8+T", "CD4+T", "ABT(ENTRY)", "DP(P)_T", "DP(Q)_T")
)Visualise the UMAP of the filtered data.
plotUMAP(sce, color_by = "anno_lvl_2_final_clean")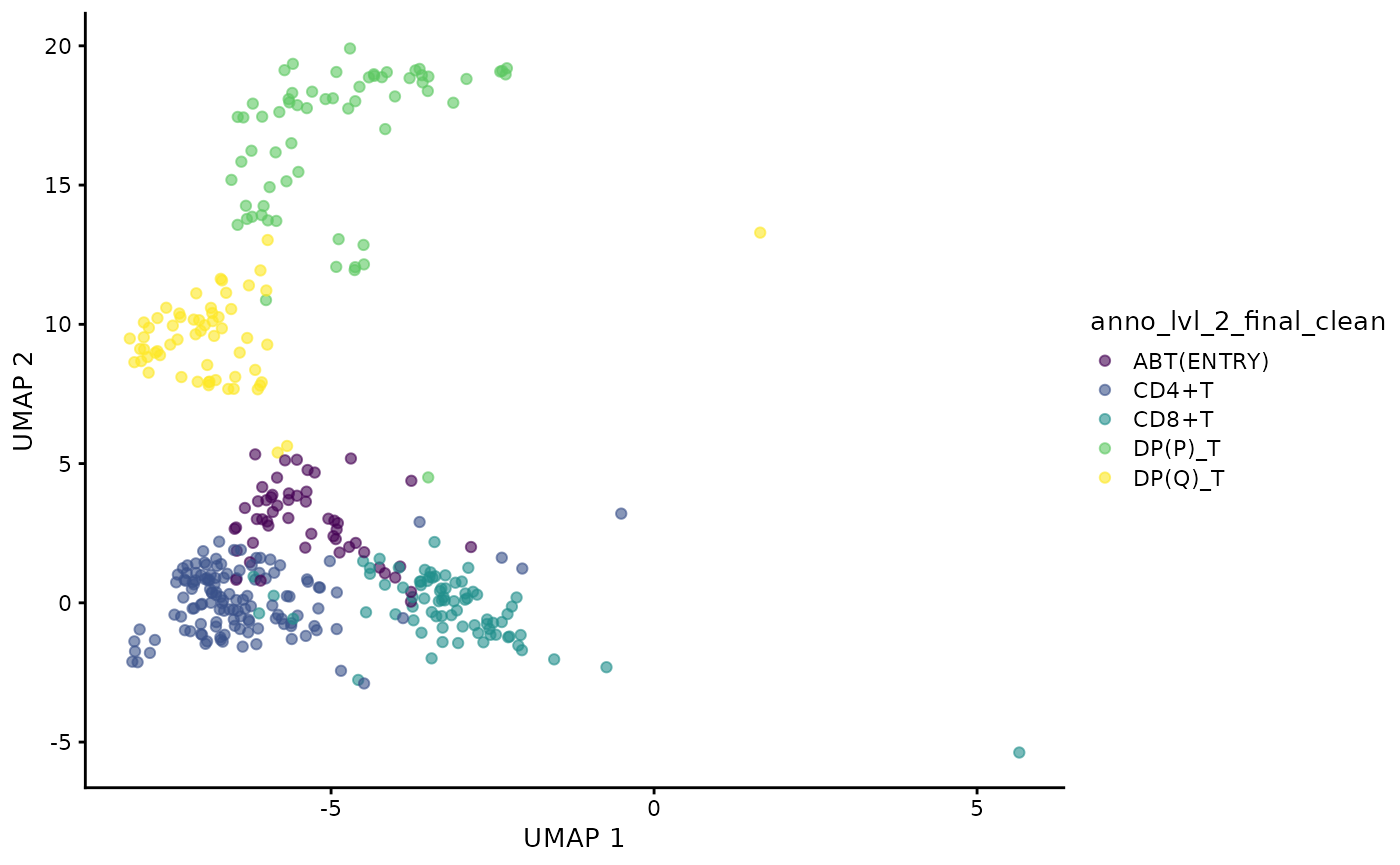
Milo object and neighbourhood graph construction
We will use miloR to create the pseudobulks based on the gene expression data. The goal is to construct a neighbourhood graph with many neighbors with which we can sample the representative neighbours to form the objects.
library(miloR)
milo_object <- Milo(sce)
milo_object <- buildGraph(milo_object, k = 30, d = 20, reduced.dim = "X_scvi")
milo_object <- makeNhoods(milo_object, reduced_dims = "X_scvi", d = 20, prop = 0.3)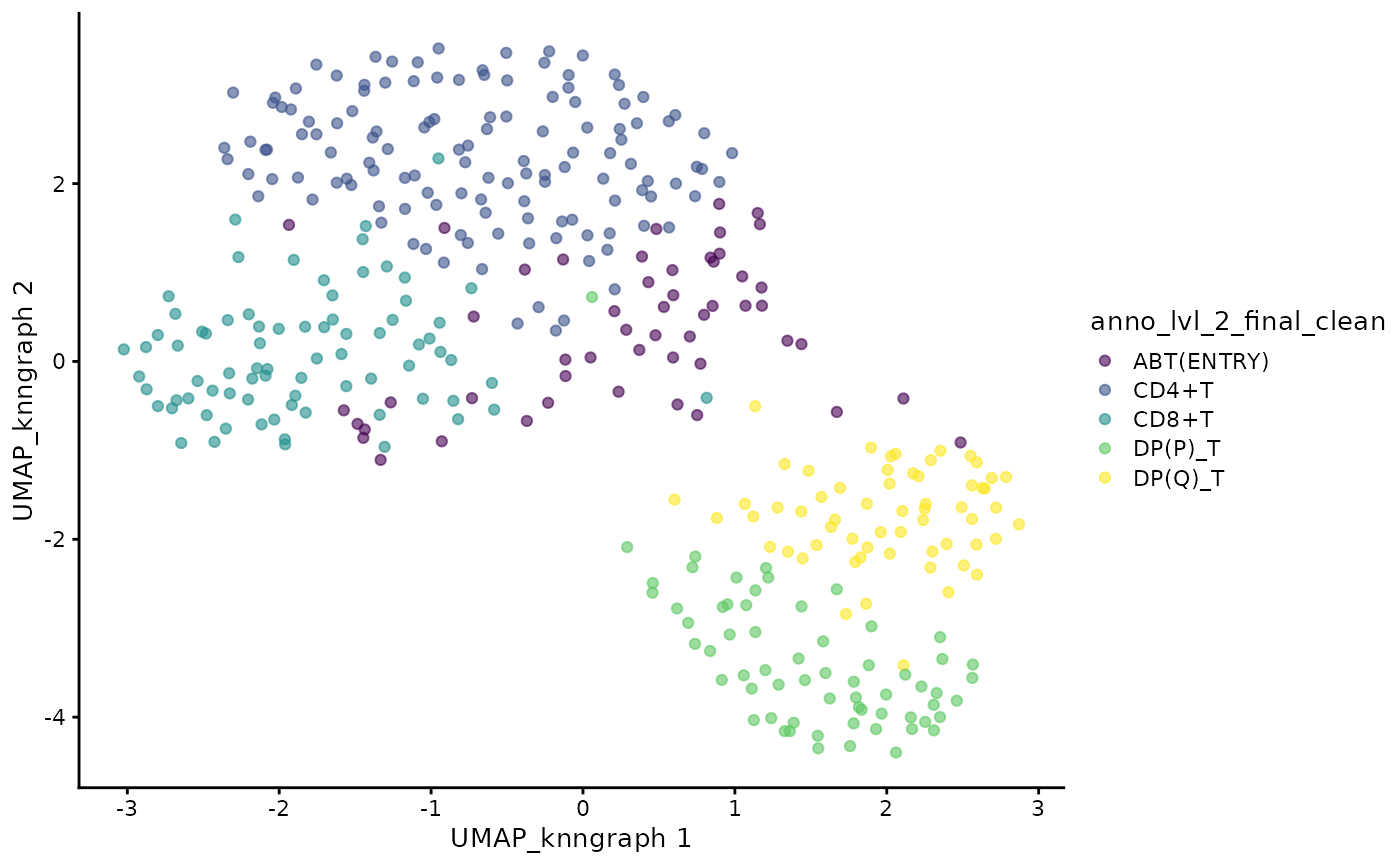
Construct pseudobulked VDJ feature space
Next, we will construct the pseudobulked VDJ feature space using the neighbourhood graph constructed above. We will also run PCA on the pseudobulked VDJ feature space.
pb.milo <- vdjPseudobulk(milo_object, mode_option = "abT", col_to_take = "anno_lvl_2_final_clean")
pb.milo <- runPCA(pb.milo, assay.type = "Feature_space", ncomponents = 20)We can visualise the PCA of the pseudobulked VDJ feature space.
plotPCA(pb.milo, color_by = "anno_lvl_2_final_clean")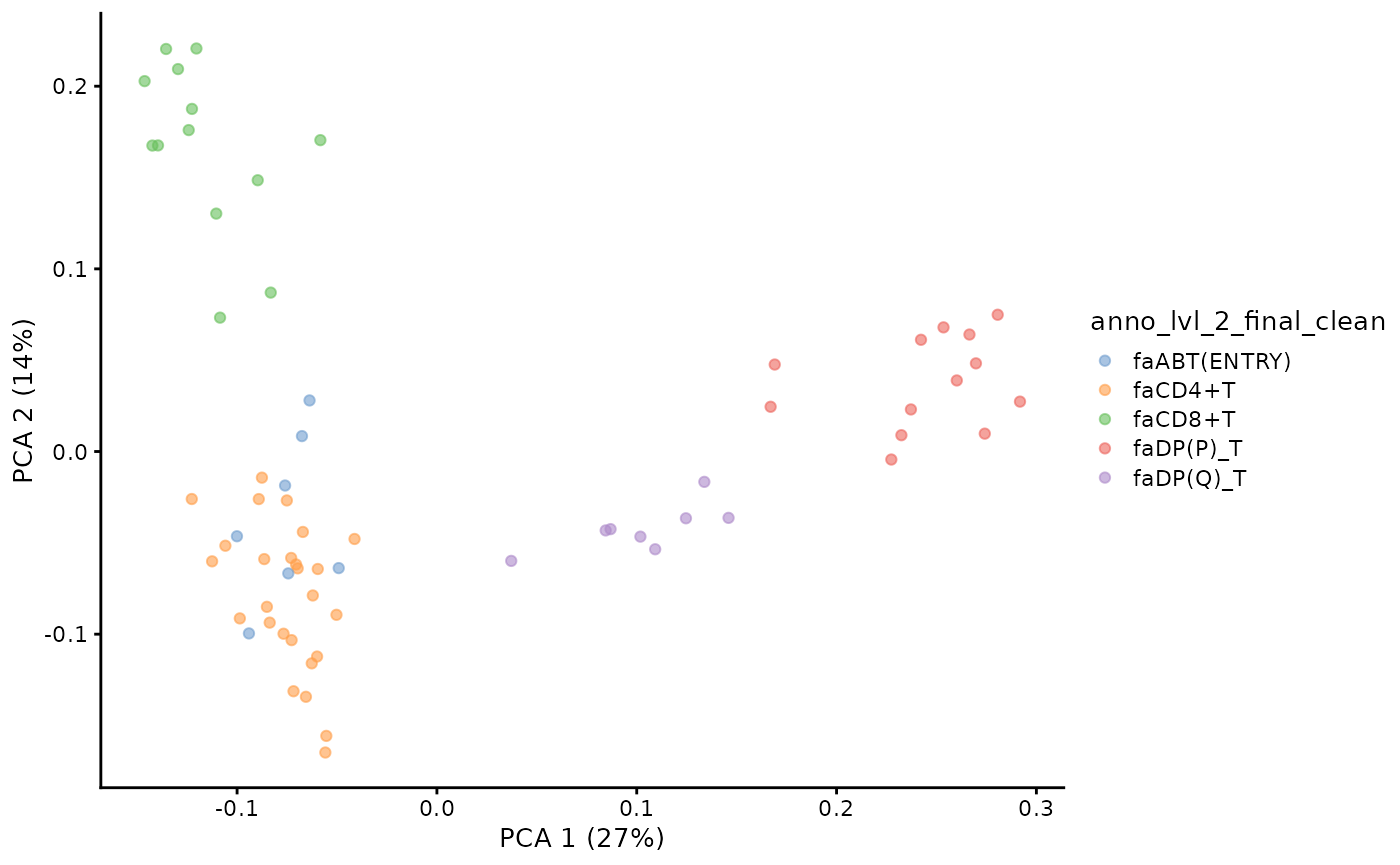
TCR trajectory inference using Absorbing Markov Chain
In the original dandelion python package, the trajectory
inference is done using the palantir package. Here, we
implement the absorbing markov chain approach in dandelionR to infer the
trajectory, leveraging on destiny for diffusion map
computation.
Define root and branch tips
# extract the PCA matrix
pca <- t(as.matrix(reducedDim(pb.milo, type = "PCA")))
# define the CD8 terminal cell as the top-most cell and CD4 terminal cell as the bottom-most cell
branch.tips <- c(which.max(pca[2, ]), which.min(pca[2, ]))
names(branch.tips) <- c("CD8+T", "CD4+T")
# define the start of our trajectory as the right-most cell
root <- which.max(pca[1, ])Construct diffusion map
library(destiny)
# Run diffusion map on the PCA
dm <- DiffusionMap(t(pca), n_pcs = 10, n_eigs = 10)Compute diffussion pseudotime on diffusion map
# the root is automatically called DPT + index of the root cell
DPTroot <- paste0("DPT", root)
# store pseudotime in milo object
pb.milo$pseudotime <- dif.pse[[DPTroot]]
# set the colours for pseudotime
pal <- colorRampPalette(rev((RColorBrewer::brewer.pal(9, "RdYlBu"))))(255)
plotPCA(pb.milo, color_by = "pseudotime") + scale_colour_gradientn(colours = pal)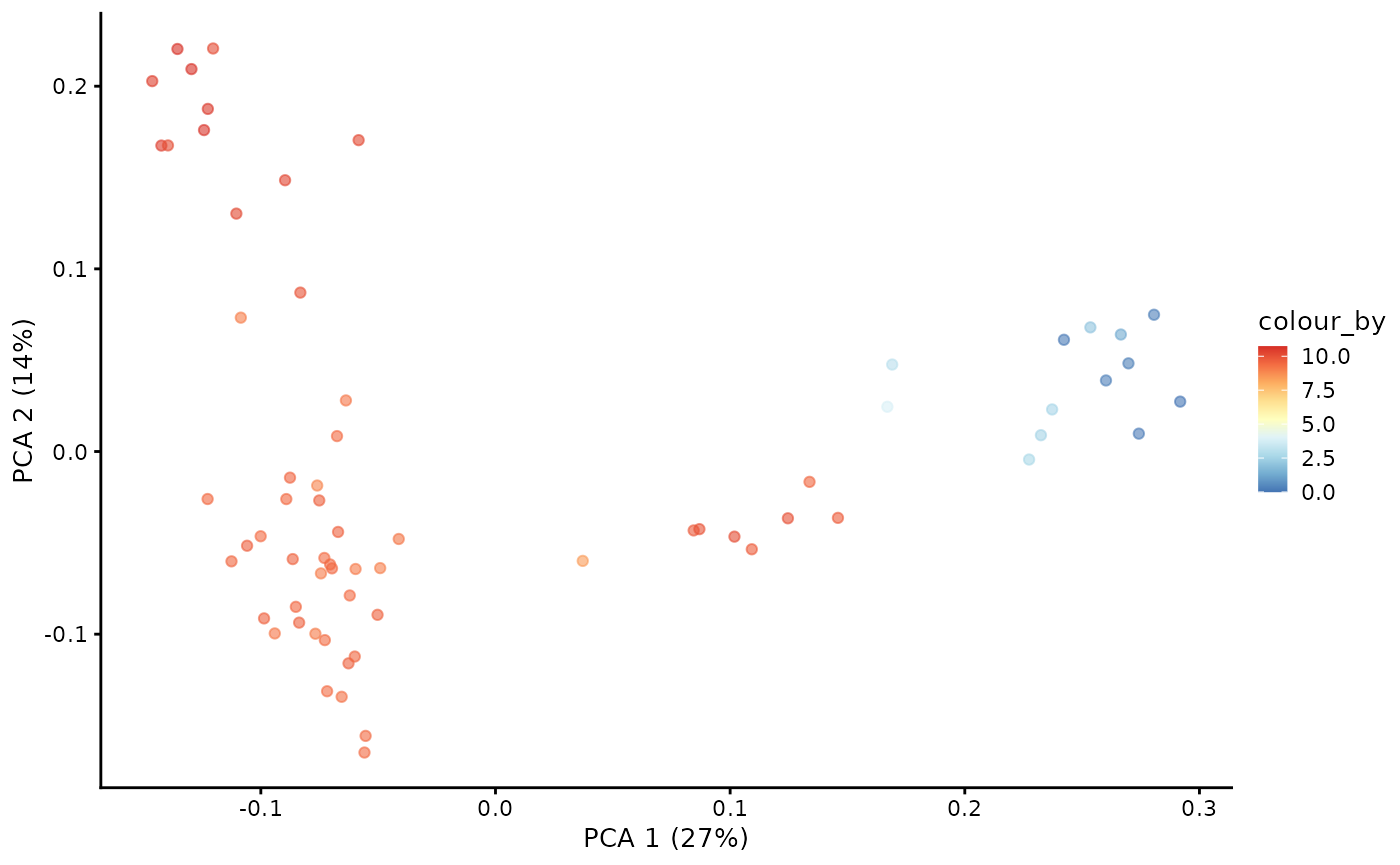
Markov chain construction on the pseudobulk VDJ feature space
pb.milo <- markovProbability(
milo = pb.milo,
diffusionmap = dm,
terminal_state = branch.tips,
root_cell = root,
pseudotime_key = "pseudotime",
knn = 30
)Visualising branch probabilities
With the Markov chain probabilities computed, we can visualise the branch probabilities towards CD4+ or CD8+ T-cell fate on the PCA plot.
plotPCA(pb.milo, color_by = "CD8+T") + scale_color_gradientn(colors = pal)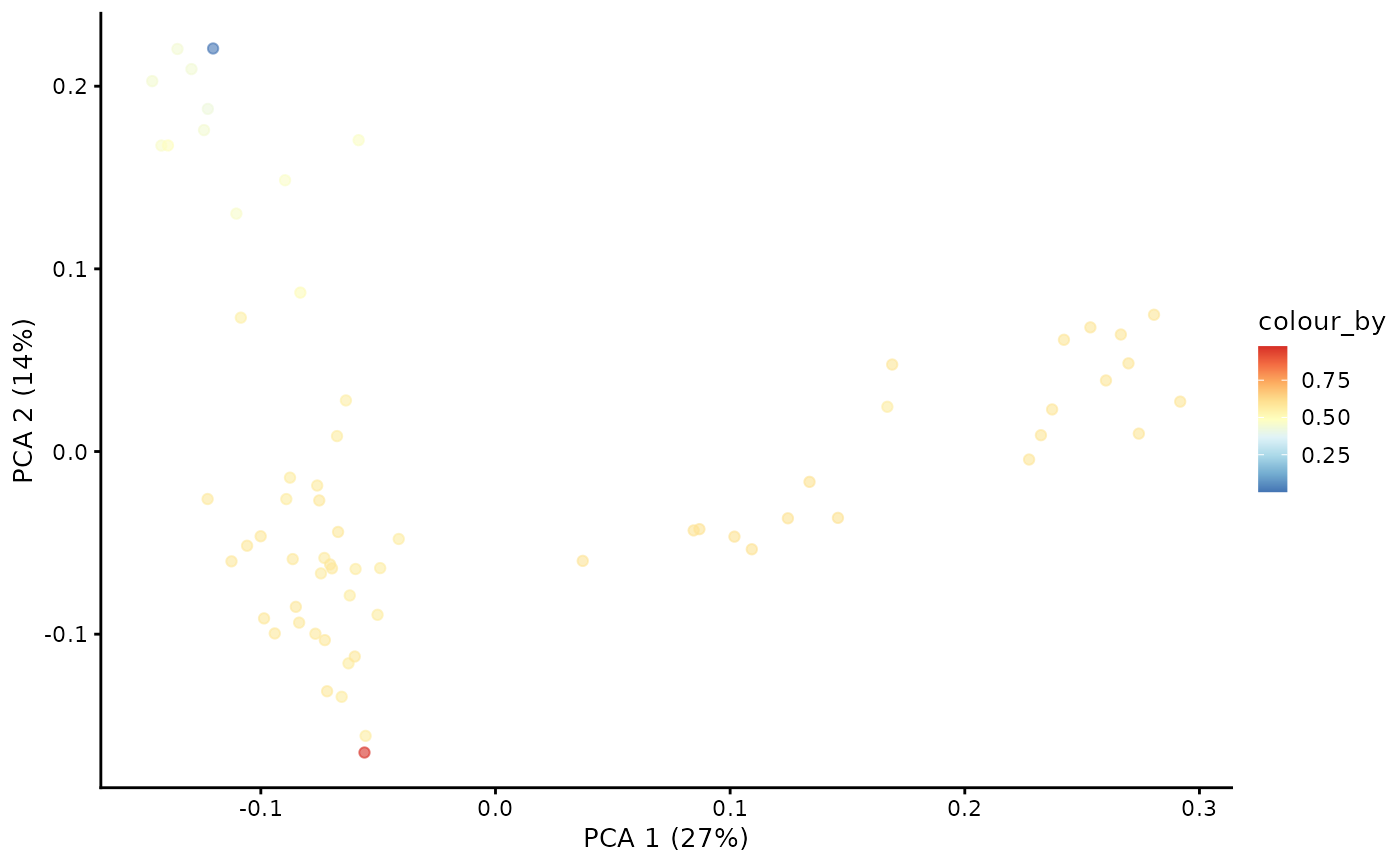
plotPCA(pb.milo, color_by = "CD4+T") + scale_color_gradientn(colors = pal)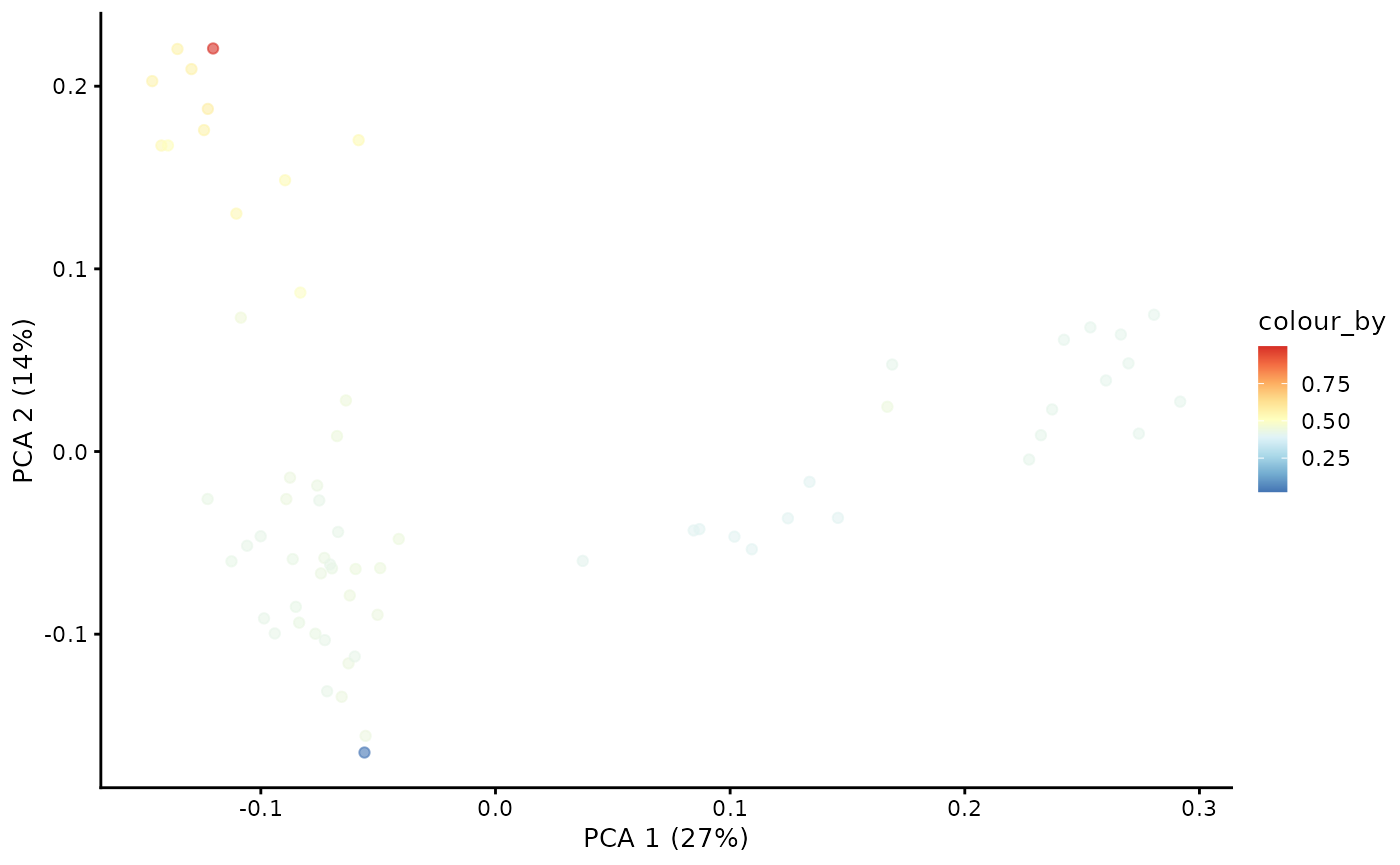
Transfer
The next step is to project the pseudotime and the branch probability information from the pseudobulks back to each cell in the dataset. If the cell do not belong to any of the pseudobulk, it will be removed. If a cell belongs to multiple pseudobulk samples, its value should be calculated as a weighted average of the corresponding values from each pseudobulk, where each weight is inverse of the size of the pseudobulk.
Project pseudobulk data to each cell
cdata <- projectPseudotimeToCell(milo_object, pb.milo, branch.tips)Visualise the trajectory data on a per cell basis
plotUMAP(cdata, color_by = "anno_lvl_2_final_clean", dimred = "UMAP_knngraph")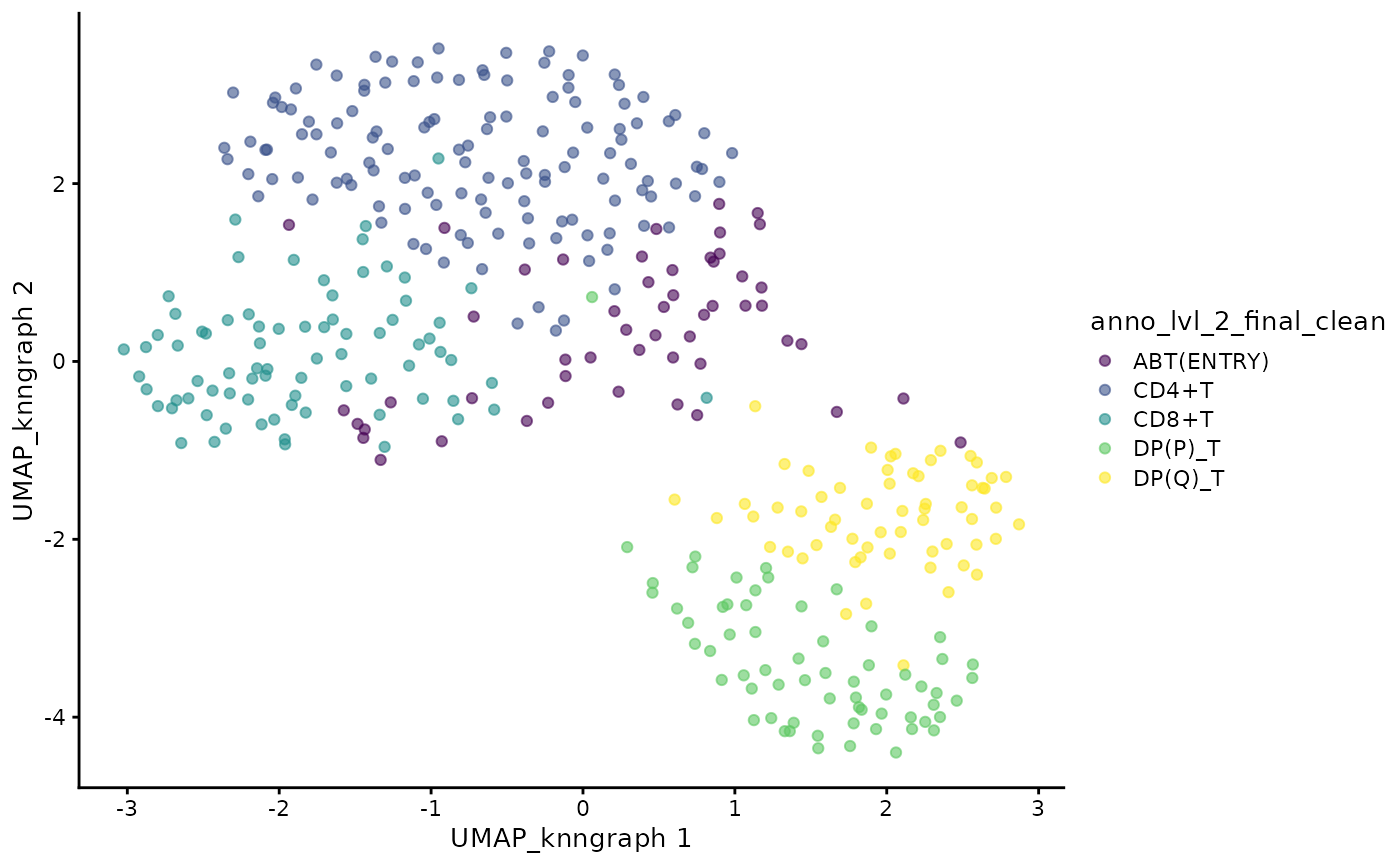
plotUMAP(cdata, color_by = "pseudotime", dimred = "UMAP_knngraph") + scale_color_gradientn(colors = pal)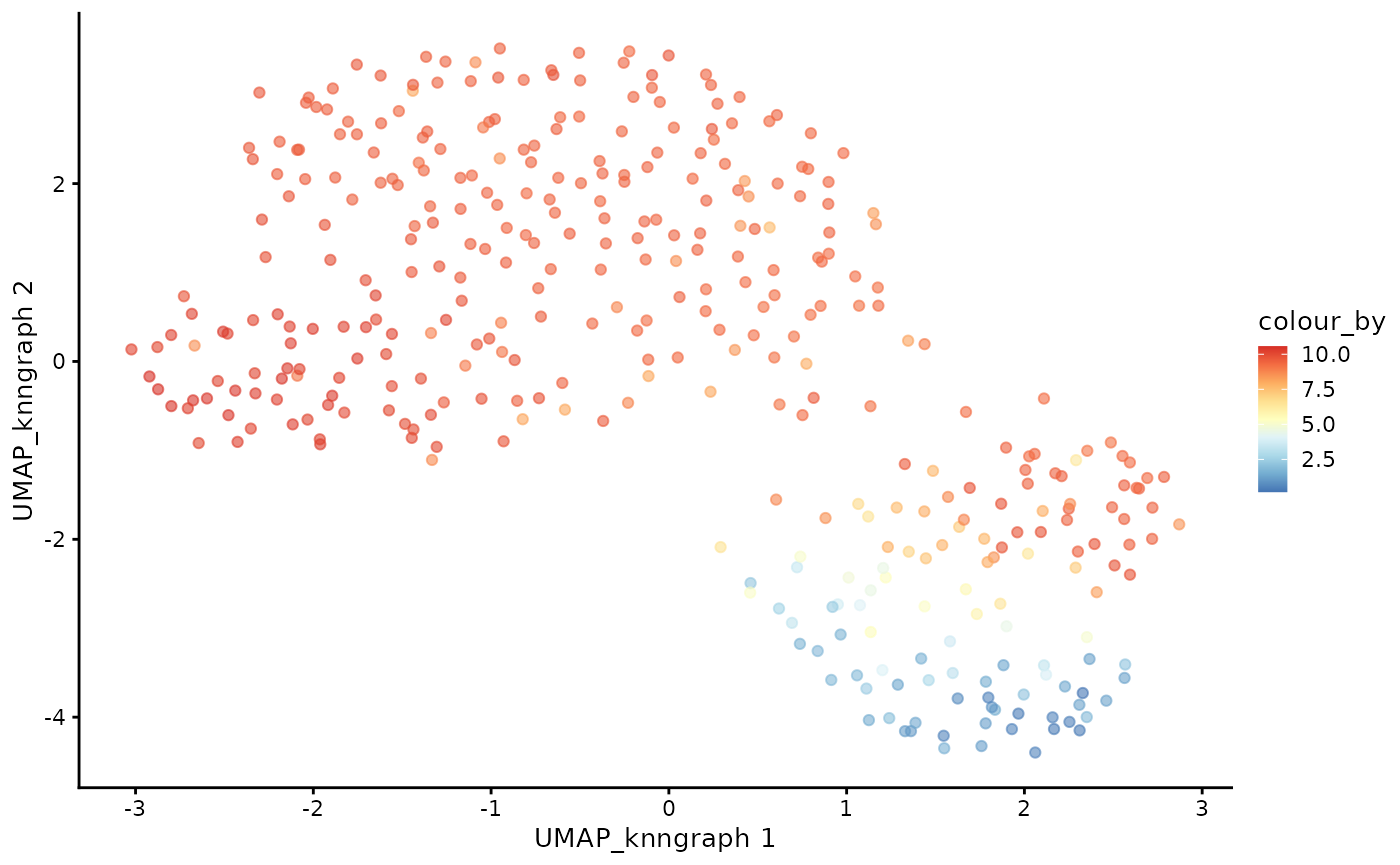
plotUMAP(cdata, color_by = "CD4+T", dimred = "UMAP_knngraph") + scale_color_gradientn(colors = pal)
plotUMAP(cdata, color_by = "CD8+T", dimred = "UMAP_knngraph") + scale_color_gradientn(colors = pal)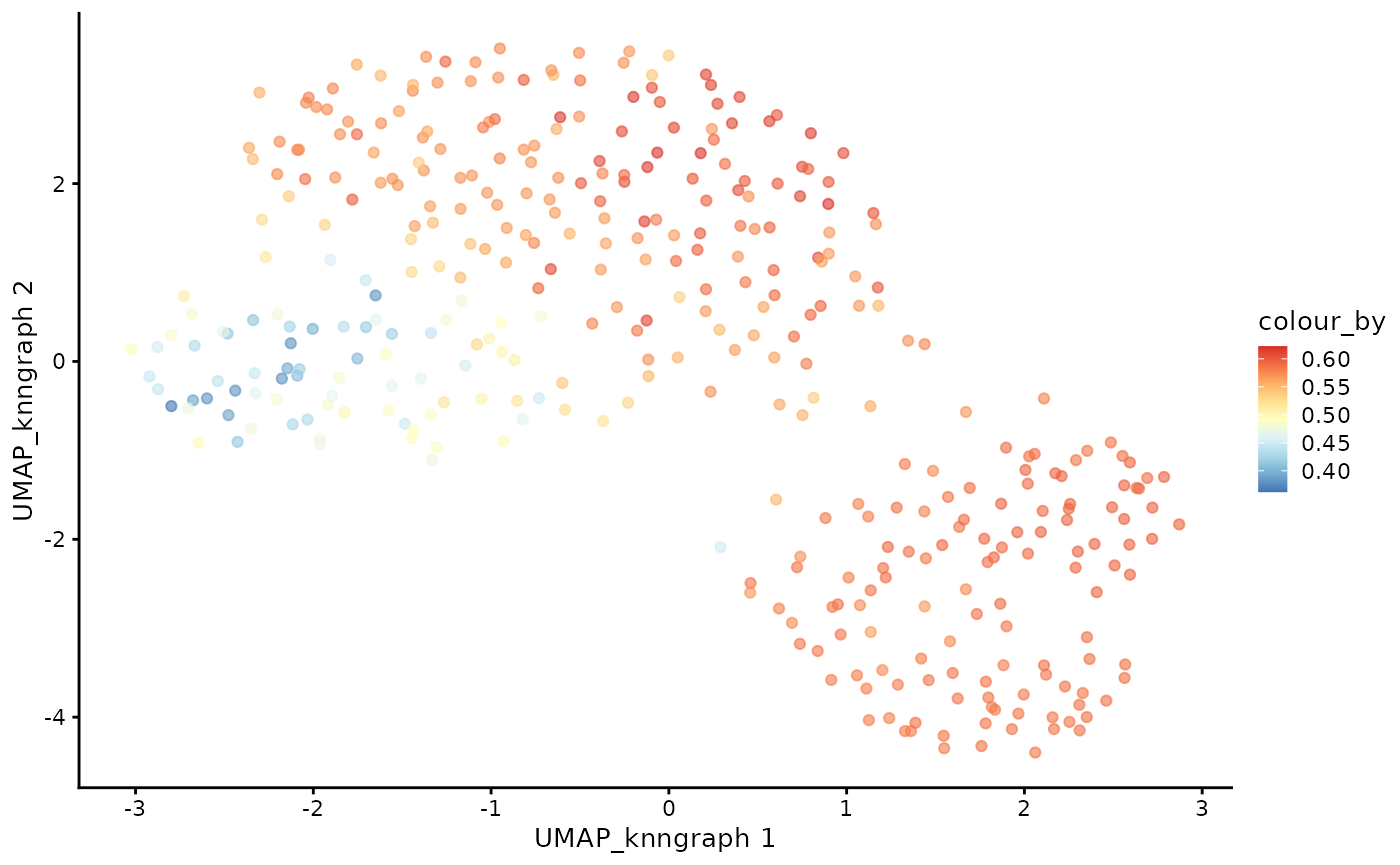
And that’s it! We have successfully inferred the trajectory of the T-cells in this dataset!
Session info
## R Under development (unstable) (2025-01-11 r87563)
## Platform: x86_64-pc-linux-gnu
## Running under: Ubuntu 24.04.1 LTS
##
## Matrix products: default
## BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
## LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
##
## locale:
## [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
## [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
## [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
## [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
##
## time zone: UTC
## tzcode source: system (glibc)
##
## attached base packages:
## [1] stats4 stats graphics grDevices utils datasets methods
## [8] base
##
## other attached packages:
## [1] miloR_2.3.0 edgeR_4.5.1
## [3] limma_3.63.3 scater_1.35.0
## [5] scuttle_1.17.0 SingleCellExperiment_1.29.1
## [7] SummarizedExperiment_1.37.0 Biobase_2.67.0
## [9] GenomicRanges_1.59.1 GenomeInfoDb_1.43.2
## [11] IRanges_2.41.2 S4Vectors_0.45.2
## [13] BiocGenerics_0.53.3 generics_0.1.3
## [15] MatrixGenerics_1.19.1 matrixStats_1.5.0
## [17] scRepertoire_2.2.1 ggplot2_3.5.1
## [19] dandelionR_0.99.0 destiny_3.21.0
## [21] BiocStyle_2.35.0
##
## loaded via a namespace (and not attached):
## [1] cubature_2.1.1 splines_4.5.0 tibble_3.2.1
## [4] polyclip_1.10-7 xts_0.14.1 lifecycle_1.0.4
## [7] globals_0.16.3 lattice_0.22-6 MASS_7.3-64
## [10] magrittr_2.0.3 vcd_1.4-13 sass_0.4.9
## [13] rmarkdown_2.29 jquerylib_0.1.4 yaml_2.3.10
## [16] spam_2.11-0 sp_2.1-4 cowplot_1.1.3
## [19] RColorBrewer_1.1-3 abind_1.4-8 purrr_1.0.2
## [22] ggraph_2.2.1 hash_2.2.6.3 nnet_7.3-20
## [25] pracma_2.4.4 tweenr_2.0.3 evmix_2.12
## [28] GenomeInfoDbData_1.2.13 ggrepel_0.9.6 irlba_2.3.5.1
## [31] listenv_0.9.1 iNEXT_3.0.1 MatrixModels_0.5-3
## [34] RSpectra_0.16-2 parallelly_1.41.0 pkgdown_2.1.1
## [37] codetools_0.2-20 smoother_1.3 DelayedArray_0.33.3
## [40] ggforce_0.4.2 tidyselect_1.2.1 UCSC.utils_1.3.0
## [43] farver_2.1.2 ScaledMatrix_1.15.0 viridis_0.6.5
## [46] jsonlite_1.8.9 BiocNeighbors_2.1.2 e1071_1.7-16
## [49] tidygraph_1.3.1 progressr_0.15.1 Formula_1.2-5
## [52] survival_3.8-3 ggalluvial_0.12.5 systemfonts_1.1.0
## [55] tools_4.5.0 ragg_1.3.3 stringdist_0.9.15
## [58] Rcpp_1.0.14 glue_1.8.0 gridExtra_2.3
## [61] SparseArray_1.7.2 laeken_0.5.3 xfun_0.50
## [64] ranger_0.17.0 TTR_0.24.4 ggthemes_5.1.0
## [67] dplyr_1.1.4 withr_3.0.2 numDeriv_2016.8-1.1
## [70] BiocManager_1.30.25 fastmap_1.2.0 boot_1.3-31
## [73] bluster_1.17.0 SparseM_1.84-2 VIM_6.2.2
## [76] digest_0.6.37 rsvd_1.0.5 R6_2.5.1
## [79] textshaping_0.4.1 colorspace_2.1-1 gtools_3.9.5
## [82] tidyr_1.3.1 hexbin_1.28.5 data.table_1.16.4
## [85] robustbase_0.99-4-1 class_7.3-23 graphlayouts_1.2.1
## [88] httr_1.4.7 htmlwidgets_1.6.4 S4Arrays_1.7.1
## [91] scatterplot3d_0.3-44 uwot_0.2.2 pkgconfig_2.0.3
## [94] gtable_0.3.6 lmtest_0.9-40 XVector_0.47.2
## [97] htmltools_0.5.8.1 carData_3.0-5 dotCall64_1.2
## [100] SeuratObject_5.0.2 scales_1.3.0 knn.covertree_1.0
## [103] ggdendro_0.2.0 knitr_1.49 rjson_0.2.23
## [106] reshape2_1.4.4 curl_6.1.0 proxy_0.4-27
## [109] cachem_1.1.0 zoo_1.8-12 stringr_1.5.1
## [112] parallel_4.5.0 vipor_0.4.7 desc_1.4.3
## [115] pillar_1.10.1 grid_4.5.0 vctrs_0.6.5
## [118] pcaMethods_1.99.0 VGAM_1.1-12 car_3.1-3
## [121] BiocSingular_1.23.0 beachmat_2.23.6 cluster_2.1.8
## [124] beeswarm_0.4.0 evaluate_1.0.3 truncdist_1.0-2
## [127] cli_3.6.3 locfit_1.5-9.10 compiler_4.5.0
## [130] rlang_1.1.4 crayon_1.5.3 future.apply_1.11.3
## [133] labeling_0.4.3 plyr_1.8.9 fs_1.6.5
## [136] ggbeeswarm_0.7.2 stringi_1.8.4 viridisLite_0.4.2
## [139] BiocParallel_1.41.0 assertthat_0.2.1 gsl_2.1-8
## [142] munsell_0.5.1 quantreg_5.99.1 Matrix_1.7-1
## [145] RcppHNSW_0.6.0 RcppEigen_0.3.4.0.2 patchwork_1.3.0
## [148] future_1.34.0 statmod_1.5.0 evd_2.3-7.1
## [151] igraph_2.1.3 memoise_2.0.1 bslib_0.8.0
## [154] DEoptimR_1.1-3-1 ggplot.multistats_1.0.1